Myasthénie

INTRODUCTION/GÉNÉRALITÉ
A) Myasthénie (ou myasthenia gravis)
- Maladie auto-immune liée à un blocage des récepteurs de la plaque motrice par des anticorps anti-récepteurs de l’acétylcholine ou d’autres types d’anticorps : c’est un bloc post-synaptique
- La responsabilité du thymus est importante : les récepteurs de l’acétylcholine des cellules myoïdes du thymus entraîneraient la stimulation d’anticorps contre les récepteurs de la jonction neuromusculaire ; le thymus serait une source de lymphocytes T helper stimulant la production de ces anticorps par les lymphocytes B ;
- Elle atteint surtout des adultes mais peut survenir à tout âge ;
- Entre 20 et 30 ans, elle est plus fréquente chez la femme que chez l’homme (dans une proportion de 3 pour 2), alors qu’au-dessus de 60 ans les cas masculins sont les plus fréquents.
B) Syndrome myasthénique de Lambert-Eaton
- Maladie auto-immune due à des autoanticorps anti-canaux calciques voltage-dépendants et à une insuffisance de libération de l’acétylcholine : c’est un bloc présynaptique
- Surtout chez le sujet de sexe masculin âgé de plus de 40 ans
- Dans 70 % des cas, syndrome paranéoplasique (+++) : 60 % de cancers intrathoraciques, 50 % de cancers bronchiques à petites cellules, 10 % de cancers différents (rein, vessie)
- Dans 15 % des cas, associé à une autre maladie auto-immune (Biermer, Sjögren…)
- Dans 15 % des cas, syndrome isolé.1
HISTORIQUE
Votre texte ici
PHYSIOPATHOLOGIE
- La physiopathologie de la myasthénie est en partie éclaircie et s’articule autour de deux acteurs essentiels : des anticorps pathogènes et le thymus. Trois quarts des patients présentant une forme généralisée de toute gravité et de tout âge et la moitié de ceux avec une forme oculaire ont des anticorps anti-RACh, dosés par une technique d’immunoprécipitation. Ces anticorps induisent une réduction du nombre RACh et, par voie de conséquence, une perte de la marge de sécurité de la transmission neuromusculaire.
- Trois modes d’action ont été décrits : blocage du site de fixation de l'acétylcholine, dégradation accélérée du RACh membranaire (modulation antigénique) et destruction par le complément de la membrane post-synaptique. Ni le taux d’anticorps anti-RACh ni le répertoire antigénique reconnu par ces anticorps ne sont corrélés à la sévérité de la maladie.
- Dans 40% des myasthénies généralisées sans anti-RACh, on détecte des anticorps dirigés contre une autre molécule post-synaptique, MuSK, qui est une tyrosine-phospho-kinase impliquée dans la transcription du RACh et dans son ancrage membranaire. Pour les myasthénies généralisées sans anticorps anti-RACh ni anti-MuSK, dites séronégatives, deux catégories d’anticorps ont été récemment décrits grâce à des techniques d’immunomarquage sur cellules HEK (Human embryonic kidney) : les anticorps anti-RACh à faible affinité et les anticorps anti LRP4. LRP4 est le récepteur de l’agrine, qui active MuSK. Le dosage de ces anticorps n’est pas encore disponible en routine, mais une équipe française est activement impliquée dans la réalisation de ce test.
- La myasthénie néonatale (MNN), syndrome myasthénique transitoire affectant 15% des nouveaux-nés de mère myasthénique à la naissance, est due à un transfert passif des anticorps maternels anti- RACh, beaucoup plus rarement anti-MuSK. Elle sera présentée plus loin.
- Connus bien avant les anticorps anti-RACh, les anticorps anti- muscles striés ont été d’abord mis en évidence en immunofluorescence puis par d’autres techniques immunologiques. Si le taux d’anticorps anti-muscles striés n’est pas corrélé à la sévérité de la maladie, il l’est clairement à la présence d’un thymome (80% des cas ont des anticorps) sauf chez les sujets âgés chez lesquels on détecte ces anticorps dans la moitié des cas, alors qu’ils n’ont pas de thymome. Les anticorps anti-titine sont également étroitement associés à la présence d’un thymome chez le patient myasthénique de moins de 60 ans.
- Le thymus est un organe lymphoïde primaire qui joue un rôle majeur chez le fœtus et dans les toutes premières années de vie, dans l’acquisition de l’immunocompétence des lymphocytes. Par la suite, le thymus s’involue. Le rôle du thymus dans la myasthénie est suggéré par le bénéfice de la thymectomie et la fréquence des anomalies histologiques. De plus, après greffe de fragments de thymus de patients myasthéniques, la souris immunodéficiente SCID développe des signes myaSthéniques et produit des anticorps anti-RACh humain. Chez 60% des patients qui appartiennent en grande majorité à la population féminine de moins de 45 ans, le thymus est le siège d'une hyperplasie caractérisée par la présence de centres germinatifs de type ganglionnaire, composés de lymphocytes B et d’une couronne de lymphocytes T. C’est dans les cas de myasthénie du sujet jeune avec hyperplasie que les taux d’anticorps anti-RACh sont les plus élevés et qu’une association avec l’haplotype HLAB8 DR3 est trouvée. Le thymus est le siège d’une hyperactivation vis-à-vis du RACh qui est exprimé au niveau des cellules épithéliales. Plusieurs cytokines et chemokines sont hyper-exprimées, participant à l’hyperactivation thymique. De plus, la fonction des lymphocytes thymiques « régulateurs» contrôlant l’activité des lymphocytes effecteurs est déficiente. Enfin, certains travaux suggèrent qu’une infection virale, et particulièrement l’EBV (Epstein Barr virus), pourrait être l’évènement déclenchant de l’emballement thymique. Au-delà de 40 ans, le thymus est involutif chez la plupart des patients.
- Quinze à vingt pour cent des patients présentent un thymome, prolifération cellulaire constituée de cellules épithéliales qui constituent le contingent tumoral et de lymphocytes, survenant habituellement après 40 ans. Le thymome se caractérise par :
- son caractère extensif au-delà de la capsule, définissant le thymome malin
- une profonde désorganisation architecturale
- une expression anormale par les cellules épithéliales néoplasiques d’antigènes du muscle strié (ex titine) et d’épitopes du RACh
- une réduction du nombre de cellules T régulatrices thymiques
- un déficit de la molécule AIRE exprimée par les cellules épithéliales et assurant la présentation aux thymocytes d’autoantigènes, dont le RACh avec pour conséquence un défaut de sélection de ces thymocytes favorisant la libération en périphérie de clones sensibilisés au RACh, qui vont précipiter la myasthénie.1
ÉPIDÉMIOLOGIE
- Elle concerne plusieurs milliers de patient en France (prévalence estimée dans la littérature de 50 à 200/million).
- Si elle débute à tout âge, de 6 mois à plus de 80 ans, la myasthénie affecte dans 60% des cas surtout des adultes jeunes, de moins de 40 ans
- En majorité des femmes.
- Dans près de la moitié des cas, les premières manifestations sont purement oculaires avec ptosis et diplopie, mais après un an d’évolution chez 80 à 90 % des patients, d’autres territoires sont affectés, muscles pharyngo-laryngés et/ou muscles des membres et/ou muscles respiratoires : la myasthénie est alors généralisée
FACTEURS DE RISQUES
Votre texte ici
EXAMEN CLINIQUE
Quelquefois évident, le diagnostic est souvent difficile et longtemps méconnu.
A) Déficit moteur variable dans le temps (fatigabilité, ou phénomène myasthénique), qui :
- apparaît ou augmente à l’effort ;
- peut se manifester dans les muscles directement mis en action au cours de l’effort ou à distance d’eux ;
- augmente en fin de journée ;
- se corrige au repos.
- ce déficit moteur est sélectif : il prédomine pour certains muscles (ci-après par ordre de fréquence).
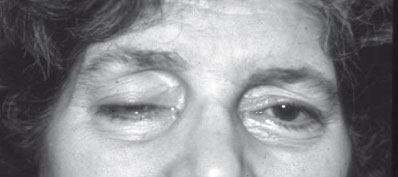
1) Muscles oculaires et palpébraux
- Ptosis unilatéral au début, qui peut se bilatéraliser par la suite ; il reste habituellement asymétrique
- Diplopie, le plus souvent intermittente.
- Ptosis et diplopie sont augmentés par la fatigue, la lumière, la fixation d’un objet.
- La musculature pupillaire est indemne.
2) Muscles d’innervation bulbaire
- Troubles de la phonation : la voix s’éteint progressivement, devient nasonnée puis inintelligible.
- Troubles de la mastication qui apparaissent au cours des repas, le sujet se trouvant parfois dans l’obligation de soutenir sa mâchoire inférieure avec sa main.
- Troubles de la déglutition donnant parfois lieu au rejet des liquides par le nez et pouvant sérieusement entraver l’alimentation.
- Une parésie faciale donnant un faciès atone est souvent associée aux troubles bulbaires.
- La fatigabilité des muscles cervicaux est à l’origine d’une chute de la tête en avant ou de douleurs cervicales liées à un phénomène de contracture.
3) Autres muscles :
- Muscles des membres : l’atteinte prédomine sur les muscles proximaux, plutôt de la ceinture scapulaire.
- Muscles axiaux : extenseurs du tronc avec camptocormie (antéflexion progressive du tronc), muscles abdominaux.
4) Muscles respiratoires :
- L’atteinte peut conduire à une décompensation ventilatoire rapide : gravité (+++).
5) Examen clinique :
- Il fait apparaître le phénomène myasthénique en utilisant des tests de répétition des mouvements, comme celui de l’abduction répétée des bras, de l’accroupissement, de l’occlusion des paupières ou de la fixation latérale prolongée du regard.
L’examen peut être normal si les symptômes sont intermittents et s’il est réalisé en période intercritique. Ceci contribue à la difficulté du diagnostic de cette maladie.
B) Formes cliniques
1) Formes oculaires de myasthénie
- Dans 15 à 20 % des cas.
- L’atteinte reste limitée aux muscles oculomoteurs tout au long de l’évolution.
- Il faut les distinguer d’une forme oculaire au début qui se généralise ensuite.
- Ces formes posent essentiellement un problème fonctionnel.
2) Formes avec anti-MuSK
- Atteinte bulbaire plus fréquente que dans les autres formes.
- Atrophie musculaire des muscles d’innervation bulbaire (langue).
- Fréquence des crises myasthéniques et gravité (++).
- Pas de pathologie thymique associée à la myasthénie avec anticorps anti-MuSK.
3) Myasthénie néonatale
- Transitoire, elle survient chez 10 à 25 % des enfants de mère myasthénique.
- Les symptômes se manifestent très précocement, durant les 24 premières heures de la vie et se prolongent 2 à 3 semaines, rarement jusqu’à 15 semaines, nécessitant un traitement anticholinestérasique puis ils régressent spontanément.
- Hypotonie associée à des troubles de la succion, de la déglutition et de la respiration
EXAMENS COMPLÉMENTAIRES
A) Recherche des autoanticorps
1) Anticorps anti-récepteurs de l’acétylcholine (anti-RAC)
- présents chez 80 % des malades avec myasthénie généralisée et chez 50 % de ceux avec myasthénie oculaire
- pas de corrélation entre le taux d’anticorps et la gravité de la maladie d’un patient à l’autre ;
- chez un même sujet, le taux peut fluctuer en fonction de l’évolutivité de la maladie ;
- dans les thymomes malins, le taux est très élevé ;
- les formes sans anticorps anti-RAC détectés peuvent être liées à d’autres types d’anticorps.
2) Anticorps anti-MuSK
- Dirigés contre une protéine associée au récepteur à l’acétylcholine : Muscle Specific Kinase
- 40 % environ des formes sans anticorps anti-RAC.
3) Formes séronégatives
- Pas d’anti-RAC et pas d’anti-MuSK.
- Il peut exister des anticorps anti-récepteurs de l’acétylcholine de faible affinité, non mis en évidence par les techniques classiques.
B) Recherche de décrément en ENMG (stimulodétection répétitive)
- Décrément : diminution de l’amplitude du potentiel évoqué musculaire (≥ 10 %) lors des stimulations répétées du nerf.
- Il apparaît pour des fréquences basses de stimulation (le plus souvent à 3 Hz) et pour la 4e stimulation.
- La sensibilité dépend du territoire où on le recherche : plus sensible dans les territoires cliniquement atteints comme un muscle proximal (trapèze) ou facial. La sensibilité diagnostique ne dépasse pas 50 à 75 %.

C) Test thérapeutique aux anticholinestérasiques
- À pratiquer en hospitalisation seulement, par crainte d’un syndrome vagotonique ou d’une crise cholinergique.
- Administration d’edrophonium IV (Enlon®, médicament en ATU) ou de néostigmine (Prostigmine®) : par exemple, 1 à 2 amp par voie SC de Prostigmine®, éventuellement associée à 0,5 mg d’atropine pour éviter les effets secondaires intestinaux et une bradycardie.
- La régression ou la disparition des signes neurologiques objectivables, ptosis en particulier, si elle est franche et rapide (délai d’action inférieur à 5 minutes pour l’edrophonium et 30 minutes pour la néostigmine) a un grand intérêt diagnostique.
D) Imagerie
- Le scanner ou l’IRM thoracique explore la loge thymique à la recherche d’un reliquat thymique suspect d’hyperplasie thymique ou d’un thymome (bénin ou malin).
DIAGNOSTICS DIFFÉRENTIELS
Autres anomalies de la transmission neuromusculaire :
A) Botulisme
- Il survient après ingestion de conserve avariée contenant une neurotoxine paralysante produite par des bactéries du genre Clostridium.
- Douze à trente-cinq heures après l’ingestion : nausées et vomissements, troubles de la vision, diplopie, sécheresse des muqueuses buccale et trachéale, déficit moteur généralisé à prédominance proximale.
- L’évolution peut être sévère en cas de survenue de complications respiratoires.
- ENMG : anomalies similaires à celles du syndrome de Lambert-Eaton, témoignant d’un trouble présynaptique de la libération d’acétylcholine.
B) Autres
- Venins de serpents.
- Intoxication par le magnésium (chez l’insuffisant rénal notamment).
- Traitement par D-pénicillamine.
- Syndromes myasthéniques congénitaux par anomalie génétique : très rares
ÉTIOLOGIE
Votre texte ici
Maladies associées
A) Pathologies associées
1) Thymus et myasthénie
- Dans 65 % des cas, hyperplasie thymique (thymus macroscopiquement normal mais caractérisé par la prolifération de follicules germinatifs à centre clair).
- Dans 15 % des cas, thymome (tumeur thymique) ; les thymomes peuvent être bénins ou malins et doivent être opérés.
2) Myasthénie et maladies auto-immunes
- Affection thyroïdienne : environ 15 % des cas.
- Autres associations dans environ 5 % des cas (polyarthrite rhumatoïde, anémie de Biermer, lupus érythémateux disséminé, …)

PRISE EN CHARGE THÉRAPEUTIQUE
A) Traitement symptomatique
- Les anticholinestérasiques prolongent l’action de l’acétylcholine au niveau de la membrane post-synaptique par blocage réversible de l’acétylcholinestérase : pyridostigmine (Mestinon®), ambénonium (Mytelase®).
- Posologie à augmenter progressivement jusqu’à la dose optimale (efficacité versus effets secondaires), à adapter à chaque patient et en fonction de son activité physique.
- Efficacité moindre voire intolérance dans les formes avec anti-MuSK.
- Durée d’action brève, de 4 à 5 heures, nécessitant des prises répétées dans la journée.
- Risque d’une crise cholinergique avec surdosage : effets muscariniques (hypersécrétion bronchique, intestinale, salivaire et sudorale) et nicotiniques (fasciculations, crampes musculaires).
B) Traitements de fond
- Corticothérapie avec possibilité d’une aggravation transitoire la première semaine.
- Immunosuppresseur : azathioprine (Imurel®) surtout, mycophénolate mofétil (Cellcept®).
- Thymectomie : effet bénéfique sur l’évolutivité de la maladie, en particulier chez le sujet jeune de moins de 40 ans porteur d’une hyperplasie thymique ; également indiquée en cas de thymome. Elle n’est pas indiquée dans les formes avec anti-MuSK.
C) Traitements des poussées évolutives
- Immunoglobulines polyvalentes IV (par exemple, 0,4 g/kg par jour pendant 5 jours).
- Alternative : échanges plasmatiques (efficacité équivalente).
D) Autres
- Tout patient doit porter sur lui une carte de myasthénie et la liste des principaux médicaments interdits.
- Prise en charge à 100 % (ALD).
- Tous les médicaments susceptibles d’altérer la transmission neuromusculaire sont contre-indiqués au cours de la myasthénie, en distinguant les contre-indications absolues et relatives et en appréciant le rapport bénéfice-risque. En cas de doute chez un patient, il ne faut pas hésiter à consulter le Vidal pour chercher d’éventuelles contre-indications ou vérifier la liste des médicaments contre-indiqués remis habituellement au patient par son médecin spécialiste.
1) Contre-indications absolues
- Aminosides, colimycine, polymyxine, telithromycine, cyclines injectables, macrolides,fluoroquinolones
- Quinines, quinidine, hydroxychloroquine, procaïnamide
- Béta-bloquants (même en collyre)
- Diphenyl-hydantoïne, trimethadione
- Dantrolène
- D-penicillamine
- Magnésium
2) Contre-indications relatives
- Curarisants: l’usage de molécules non dépolarisantes de dégradation rapide, comme
- l’atracurium, est possible, nécessité d’un monitorage précis
- Benzodiazépines
- Neuroleptiques (phenothiazine)
- Carbamazépine
- Lithium
3) Cas particuliers
- L’allopurinol potentialise l'effet de l'azathioprine: il faut réduire la dose des 2/3 et surveiller étroitement la NFS.
- L’injection d’iode pour examen radiologique de contraste peut induire une décompensation aigüe. Elle est déconseillée en cas de poussée.
- Vaccinations : le retentissement sur la myasthénie est mal documenté. La vaccination contre la poliomyélite, le tétanos et la grippe n’entraîne pas d’aggravation lorsque la myasthénie est bien contrôlée. Les vaccins vivants (par exemple polio buccal) sont formellement contre-indiqués chez les patients sous corticoïdes ou immunosuppresseurs.
- L’interféron alpha et bêta peuvent aggraver voire induire une myasthénie.
- L’utilisation de patch de nicotine pour le sevrage de l’intoxication tabagique peut aggraver la myasthénie.
- Toxine botulique à des fins esthétiques contre-indiquée
- Statines, effet négatif dans la myasthénie rapporté mais discuté.
ÉVOLUTION/PRONOSTIC
- Chronique et capricieuse, par succession irrégulière de poussées et de rémissions, difficile à schématiser et à prévoir.
- Risques vitaux possibles, du fait des crises myasthéniques se manifestant par des troubles respiratoires, avec dyspnée et encombrement. L’évolution fatale se produit dans plus de deux tiers des cas malgré la réanimation.
- Effet de la grossesse variable mais risque d’aggravation de 30 % en post-partum.
PRÉVENTION
Votre texte ici
SURVEILLANCE
Votre texte ici
CAS PARTICULIERS
A) Syndrome myasthénique de Lambert-Eaton
- Déficit moteur des membres inférieurs accompagné d’une fatigabilité excessive, sans amyotrophie.
- Réflexes faibles ou absents, surtout aux membres inférieurs (ils sont facilités et apparaissent si on les recherche immédiatement après un effort musculaire).
- Des signes d’atteinte céphalique évoquant une myasthénie sont parfois présents (moins fréquents que dans la myasthénie).
- Dysautonomie cholinergique possible (troubles de la motricité pupillaire, de la sudation, des sécrétions salivaire et lacrymale, impuissance…).
- ENMG : potentiel d’action musculaire au repos d’amplitude très diminuée par rapport à la normale et dont l’amplitude augmente de plus de 100 % immédiatement après tétanisation ; stimulation nerveuse répétitive à fréquence rapide (10 à 50 Hz) : incrément toujours supérieur à 100 %.
- Anticorps dirigés contre les canaux calcium présynaptiques voltage-dépendants.
- Médicaments pour augmenter le nombre de quanta d’acétylcholine libérés : 3,4-diaminopyridine.
- Dans les formes paranéoplasiques, l’évolution dépend du traitement du cancer. Dans les autres formes, un traitement immunosuppresseur ou immunomodulateur peut être proposé comme dans la myasthénie.1
B) Myasthénie du sujet âgé
- La myasthénie n’est pas rare après 60 ans (late onset myasthenia gravis)
- Un début très tardif, au-delà de 80 ans, est possible (Very late onset myasthenia gravis).
- L’errance diagnostique est fréquente car le diagnostic de myasthénie n’est pas évoqué à cet âge, l’accident vasculaire étant la première hypothèse.
- Si l’expression clinique et les tests diagnostiques sont très proches de la myasthénie classique, la forme tardive se caractérise par certaines particularités :
- prédominance masculine,
- plus grande fréquence des formes oculaires pures,
- sévérité plus grande pour les formes généralisées du fait d’une composante bulbaire franche,
- risque de thymome important à la cinquantaine, devenant exceptionnel après 70 ans et, en l’absence de thymome, involution thymique associée significativement à l’HLAB7.
- Pour ce qui concerne la thérapeutique, la stratégie et la réponse sont identiques à celles du sujet plus jeune, hormis l’absence d’indication de la thymectomie en dehors du thymome.
- Ces patients sont plus exposés, du fait d’une consommation médicamenteuse élevée, à un risque d’associations médicamenteuses délétères.1
C) Myasthénie et grossesse, myasthénie néonatale, myasthénie fœtale.
- La fertilité n'est pas affectée par la maladie, c’est pourquoi la grossesse n’est pas exceptionnelle dans la myasthénie.
- Au cours de la grossesse, il y a un risque sérieux d’exacerbation des symptômes myasthéniques dans 30 à 40% des cas,surtout dans les trois premiers mois et encore plus dans les jours et premières semaines qui suivent l’accouchement.
- Entre 10 et 20% des nouveaux nés de mère myasthénique présentent une myasthénie néonatale (MNN), qui est un syndrome myasthénique transitoire dû à un transfert transplacentaire passif des anticorps maternels anti-RACh, beaucoup plus rarement anti-MuSK. Les symptômes se manifestent chez le nouveau-né précocement, durant les 24 premières heures, parfois un peu plus tardivement (jusqu’au 3ème jour) et persistent en général 2 à 3 semaines, au maximum 3 mois, puis régressent spontanément sans séquelles. La MNN se manifeste habituellement par une hypotonie, des troubles de succion et de déglutition, une faiblesse du cri, tous symptômes améliorés par les anticholinestérasiques. Le risque de fausse route et de détresse respiratoire doit être dépisté et impose le transfert en réanimation pour assurer une nutrition par sonde et une assistance ventilatoire. Aucune corrélation ne peut être établie entre la gravité de la myasthénie maternelle et la survenue ou la sévérité de la myasthénie néonatale. Selon certains auteurs le risque de MNN serait accru si les anticorps taux d’anti-RACh maternel est élevé, mais pour d’autres, il n’y aurait pas de corrélation. Il est donc conseillé d’accoucher dans une structure permettant la prise en charge de la mère et de l’enfant en réanimation (maternité de niveau 3).
- Une forme fœtale de myasthénie transmise, beaucoup plus rare, se caractérise par un début anténatal avec hydramnios, arthrogrypose témoignant de l’immobilité foetale (signes absents dans la forme classique), un tableau sévère à la naissance, une atteinte myopathique persistante (déficit facial, voix nasonnée, troubles de déglutition, faiblesse musculaire). La myasthénie foetale est due à des anticorps dirigés contre la forme foetale du RACh exprimant de la sous unité gamma à la place de la sous unité adulte, epsilon. De ce fait, la mère, épargnée par les anticorps fœtaux, est peu atteinte, voire complètement asymptomatique, en contraste avec la gravité de l’atteinte de son nouveau-né.
- Plus récemment ont été rapportées des formes foetales sans signes anténataux, prises initialement pour une forme classique de myasthénie transitoire mais qui se singularisent par une atteinte séquellaire faciale et bulbaire indiquant un début fœtal passé inaperçu. Le risque de récidive de la myasthénie transmise est important, en particulier dans les formes fœtales.
- Il a été démontré que la réduction du taux des anticorps maternels anti-RACh fœtaux, obtenue en traitant la mère pendant la grossesse par échanges plasmatiques ou immunoglobulines polyvalentes IV et/ou corticoïdes pouvait réduire la gravité du tableau de l’enfant suivant exposé à un très fort risque de récurrence.
THÉRAPIES FUTURES
Votre texte ici