Anomalie De L'intervalle QT

Mise à jour
09/08/2020
INTRODUCTION
Votre texte ici
INTERVALLE QT COURT
A) Généralités
- Le diagnostic repose essentiellement sur la mesure de l’intervalle QTc qui est raccourci. Un système de score a été proposé (Gollob et al) mais on lui préfère actuellement simplement la définition basée sur le QT corrigé (ref guidelines 2015)1
- QTcorrigé < 340 ms
- ou QTcorrigé < 360 ms avec une mutation considérée comme pathogène et/ou une histoire familiale de SQTC, et/ou une histoire familiale de mort subite < 40 ans et/ou une histoire personnelle de TV/VF en l’absence de cardiopathie.
- Un intervalle QTc court expose à un risque accru de syncope, fibrillation atriale ou ou de mort subite secondaires à des troubles du rythme ventriculaire graves à type de tachycardies ventriculaires polymorphes ou de fibrillation ventriculaire.1 Ce risque qui augmente avec le raccourcissement du QT doit être nuancé en fonction d’autres paramètres.1
- Le raccourcissement du QT est généralement acquis, en rapport avec une imprégnation en digitalique, hypercalcémie, hyperkaliémie, acidose ou hyperthermie. Il peut s’agir de façon exceptionnelle d’un syndrome du QT court congénital.1
B) Syndrome du QT court congénital
1) Étiologie
- Il s’agit d’une « canalopathie » en rapport avec une mutation de gènes intervenant dans l’électro-physiologie cellulaire.1
- Intervalle QT court d’origine génétique à transmission autosomique dominante.
- À ce jour, des mutations dans 3 gènes, KCNH2, KCNQ1 et KCNJ2, tous codant des canaux potassiques cardiaques impliqués dans la repolarisation des cellules cardiaques, ont été trouvées associées à ce syndrome.
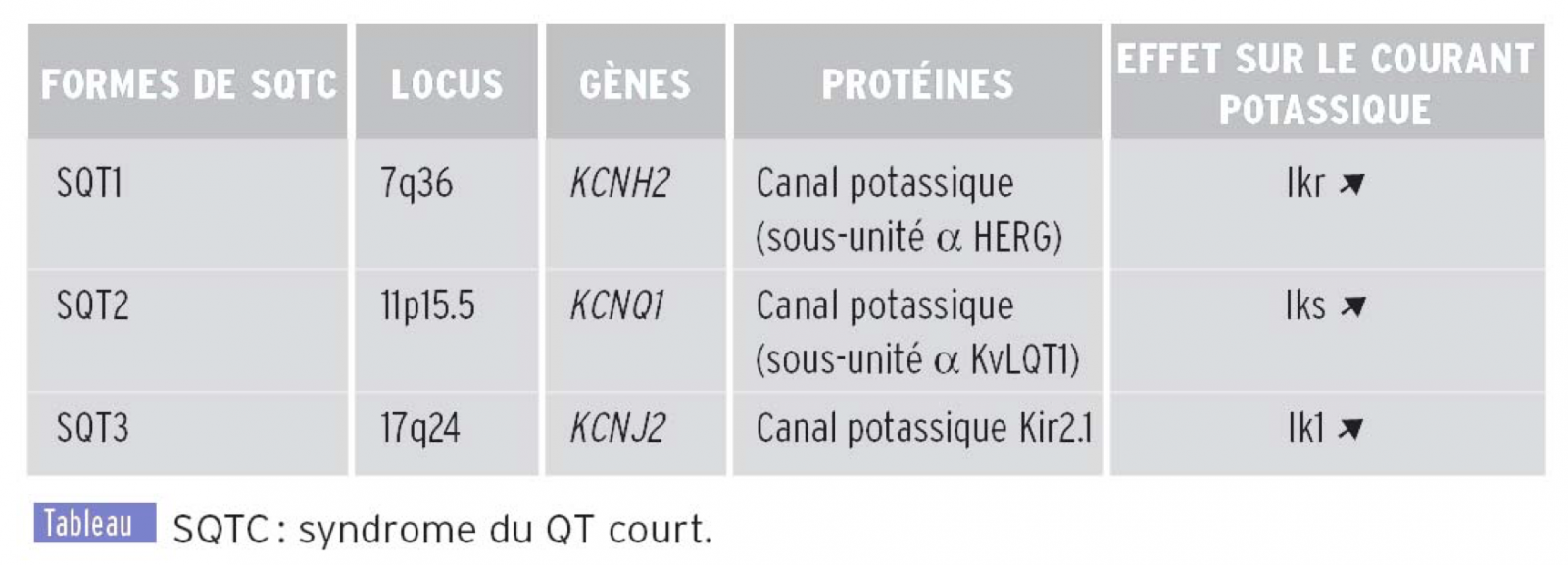
2) Épidémiologie
- Il n’y a pas de facteur déclenchant prédominant, les accidents survenant au repos ou à l’effort.
- La prévalence est inconnue et ce syndrome reste exceptionnel, la fréquence de QT courts dans les grandes cohortes de sujets sains restant en outre très rare.
- On note plus de cas de QT court chez les hommes que chez les femmes dans les registres (mais le risque d’accident rythmique semble similaire).
- Les accidents rythmiques peuvent survenir à n’importe quel âge, même chez les nouveau-nés ou nourrissons.1
3) Manifestations cliniques
- Elles sont très variables, allant de l’absence de symptôme jusqu’à des épisodes de syncopes ou des morts subites par trouble du rythme ventriculaire.1
- On note également une incidence élevée (70% des cas) de fibrillation auriculaire permanente ou paroxystique. Dans plus de la moitié des cas, la fibrillation auriculaire est la première manifestation du syndrome, avec un âge de survenue de 40 ±19 ans. Ainsi, devant une fibrillation auriculaire idiopathique du sujet jeune, il faut penser à éliminer le diagnostic de syndrome du QT court. Il existe souvent des antécédents familiaux de mort subite ou de fibrillation auriculaire touchant plusieurs générations.1
4) Examens complémentaires
a) ECG 12 dérivations
- Mesure de l’intervalle QT puis calcul de l’intervalle QT corrigé (QTc) selon la formule de Bazett (QT corrigé= QT/√RR) (ou RR représente l’intervalle entre deux QRS en sec) (en évitant les fréquences trop élevées ou trop basses).
- Les valeurs du QTc sont souvent faibles (ex. 250 ± 15 ms).1
- L’intervalle QT s’adapte mal à la fréquence cardiaque et reste constamment court (risque de faux négatif en cas de mesure lors d’une fréquence > 80/mn).
- Les ondes T sont pointues, amples et symétriques dans les dérivations précordiales, mais leur aspect varie selon le génotype responsable.
b) Holter :
- Mesure de l’intervalle QT à 60/mn
- Absence d’allongement net lors des phases lentes
c) Epreuve d’effort : QT très court à l’effort mais adaptation QT à la fréquence (pente QT/RR) réduite
d) Echocardiographie : normalité des structures cardiaques
e) Test pharmacologique : aucun (sauf ajmaline pour dépister un éventuel syndrome de Brugada associé)
f) Exploration électro-physiologique :
- Sans intérêt (pas de rôle pronostic du déclenchement d’arythmies) sauf pour mesurer les périodes réfractaires (courtes en cas de QT court, souvent < 200 ms) en cas de doute diagnostique.
- L’exploration électrophysiologique révèle des périodes réfractaires atriales et ventriculaires courtes et des arythmies ventriculaires sont fréquemment déclenchées par la stimulation ventriculaire
g) Bilan génétique :
- Indiqué pour l’instant à titre de recherche seulement.
- Nécessite une consultation de cardiogénétique avec prélèvements réalisés dans l’un des Centres de Référence, ou l’un des Centre de Compétence.
5) Traitements 1
a) Défibrillateur automatique implantable :
- Il est recommandé chez les sujets ayant présenté des arythmies ventriculaires malignes (TV, VF) et en réglant les paramètres de détection de manière à éviter les doubles comptages du QRS et de l’onde T (ample en général).
- En cas d’histoire familiale de mort subite liée au SQTC chez un patient par ailleurs asymptomatique, la quinidine est proposée, puis si celle-ci est inefficace sur le QT, il peut y avoir un place pour une discussion collégiale, notamment au sein de la filière Cardiogène, pour le défibrillateur prophylactique.
- Après stratification pronostique, un défibrillateur peut être proposé.
- Des recommandations USA 2017 sont téléchargeables en ligne.1
b) Traitement médicamenteux :
- Seule la quinidine ou le disopyramide ont pu montrer un intérêt surtout en cas de mutations sur HERG (SQTC de type 1) mais aussi dans les autres types ou sans mutation retrouvée quoique peut etre de manière moindre , sous condition d’allonger le QT et les périodes réfactaires.
- Les autres anti-arythmiques semblent inefficaces (meme si le sotalol est proposé aussi dans les recommendations, mais semble décevant dans les études). Le traitement par quinidine doit être surveillé pour dépister un allongement excessif du QT ou un effet pro-arythmique. Aucune expérience n’est disponible avec les beta-bloqueurs (qui ne modifient pas le QT) et très peu avec l’amiodarone (qui ne paraît pas toujours efficace).
- Ces traitements peuvent être proposés aux sujets avec arythmies ventriculaires malignes en cas de contre-indication (enfants) ou refus du défibrillateur, et aussi chez les asymptomatiques avec histoire de mort subite familiale. Ces traitements peuvent aussi être prescrits pour la fibrillation atriale, comme la propafénone aussi.
- L’isoprénaline a pu se montrer efficace en cas d’orage rythmique sur un cas.
- Il n’y a pas de médication contre-indiquée connue.
c) Sports de compétition :
- Ils sont contre-indiqués en l’absence de données sur le risque d’évènements lors de l’activité sportive (des accidents ont été décrits pendant l’activité physique).
d) Modalités de surveillance des patients
- ECG, Holter, épreuve d’effort : une fois par an.
- Permet de surveiller : la clinique, l’absence d’arythmies atriales ou ventriculaires et la durée du QTc
e) Conseil génétique et test génétique
- Doit être effectué au sein d’une équipe pluri-disciplinaire experte (centre de référence, centre de compétence).
- Ce conseil génétique est essentiellement fondé sur le phénotype (présence du QT court).
- Un test génétique peut être réalisé au sein d’une équipe experte avec une interprétation prudente.
f) Surveillance cardiologique de la famille
- Informer le patient de la nécessité légale d’informer (lui-même ou via le médecin après autorisation écrite) les apparentés directs (parents, fratrie et descendance) de l’existence du syndrome du QT court. Il s’agit d’une affection mal connue mais qui semble répondre à une transmission autosomique dominante (risque de transmission est de 50 % à chaque grossesse). La pénétrance est inconnue.
- Bilan cardiaque préconisé chez apparentés: ECG, +/- Holter, test d’effort en cas de doute diagnostique. Du fait de possibles formes pédiatriques très précoces, il semble prudent d’envisager ce bilan dès la naissance (simple ECG pour les nouveau-nés, puis bilan plus complet plus tard dans l’enfance).